Anemia mediterranea
Che cos'è l'anemia mediterranea (beta talassemie)?
Con il termine anemia mediterranea si indica il gruppo di malattie del sangue note come beta talassemie.
La parola talassemia deriva dal greco thalassa (mare) e haima (sangue).
Le beta talassemia sono dovute a una ridotta o mancata produzione delle globine β (due copie delle quali sono presenti insieme a due globine α in ogni molecola di emoglobina) dando luogo a ridotta produzione di globuli rossi e quindi ad anemia.
Il quantitativo di globine β mancanti viene rimpiazzato da globine α. L'eccesso di globine α può dare luogo alla produzione di precipitati che alterano la forma dei globuli rossi e ne provocano una morte prematura.
A seconda della gravità di distinguono tre tipi principali di talassemia:
- major o morbo di Cooley;
- intermedia;
- minor.
La talassemia minor coincide con la condizione di eterozigote o portatore sano.
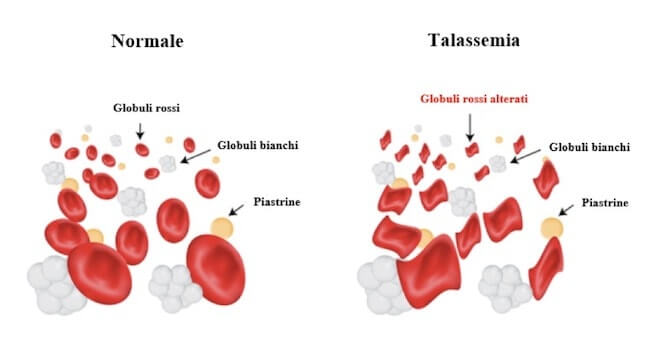
L'eccesso di globine α in sostituzione delle β mancanti può creare dei precipitati che alterano la forma dei globuli rossi compromettendone la funzionalità e provocandone una morte prematura.
Epidemiologia dell'anemia mediterranea
L'incidenza annuale della talassemia è di 1 su 100000 in tutto il mondo e di 1 su 10000 nell'Unione Europea.
Si stima che l'1,5% della popolazione (circa 80-90 milioni di persone) sia affetto da una delle tre forme di talassemia.
Le aree più colpite sono i Paesi Mediterranei, l'Asia centrale e sud-orientale, l'India, il sud della Cina, la costa del nord Africa e il Sud America.
Le percentuali più alte di portatori si riscontrano a Cipro (14%), in Sardegna (10,3%) e nel Sud-Est asiatico.
I flussi migratori volontari o forzati e i matrimoni tra individui appartenenti a diversi gruppi etnici hanno tuttavia contribuito a diffondere l'anemia mediterranea in tutto il mondo, come nel Nord Europa, dove prima era assente.
Come per l'anemia falciforme, le aree più colpite da beta talassemia sono anche quelle attualmente o in passato endemiche della malaria, secondo quanto descritto dall'"ipotesi malaria", che intravedrebbe nelle anomalie strutturali e funzionali dei globuli rossi un fattore protettivo nei confronti dell'infezione da Plasmodium falciparum.
Talassemia major
I sintomi della talassemia major si manifestano entro i due anni di vita con una grave anemia che richiede il ricorso alla trasfusione di globuli rossi.
Nei soggetti non opportunamente trattati si osservano ritardo della crescita, pallore, ittero, muscolatura poco sviluppata, continua stanchezza, irritabilità, scarso appetito, ingrossamento del fegato e della milza, ulcere alle gambe, deformazione delle ossa (tipicamente delle gambe e del cranio) a causa dell'espansione del midollo osseo.
Le regolari trasfusioni, d'altra parte, provocano un accumulo di ferro da cui derivano disfunzioni del sistema endocrino (ritardo della crescita, pubertà ritardata, diabete, insufficienza di ipofisi, tiroide, paratiroidi e delle ghiandole surrenali), cardiomiopatia dilatativa, fibrosi del fegato e cirrosi.
Frequenza ottimale delle trasfusioni e uso di agenti chelanti del ferro possono garantire la sopravvivenza fino ai 40 anni di età e un buon stile di vita.
Nel 71% dei casi la morte sopraggiunge per complicazioni cardiache dovute alla siderosi, che nonostante l'uso di chelanti non viene contrastata in modo efficace a lungo termine.
Talassemia intermedia
La talassemia intermedia si presenta tra i 2 e i 6 anni con una moderata anemia che non richiede trasfusioni regolari, ma che provoca anche in questo caso un rallentamento della crescita e dello sviluppo.
La manifestazione tipica della talassemia intermedia è l'ipertrofia del midollo osseo con conseguenti osteoporosi, deformazione e possibili fratture delle ossa e complicanze a livello di fegato, milza, linfonodi, colonna vertebrale.
Per contrastare l'iperattività del midollo osseo, ai soggetti affetti da talassemia intermedia si consiglia l'assunzione di integratori di acido folico.
Gli individui affetti da talassemia intermedia hanno inoltre un elevato rischio di ulcere alle gambe, calcoli biliari e trombosi.
Talassemia minor
La talassemia minor è in genere asintomatica o può provocare una lieve anemia.
Geneticamente è associata a una condizione di eterozigosi o di portatore sano in cui del gene della globina β sono presenti un allele normale e uno mutato.
Cause genetiche dell'anemia mediterranea
Le beta talassemie sono dovute a mutazioni puntiformi o più raramente a delezioni del gene per la globina β, situato sul cromosoma 11, dando luogo alla mancata (talassemia β0- beta zero) o alla ridotta (talassemia β+- beta più) sintesi della globina β.
La trasmissione delle mutazioni è di tipo autosomico recessivo, ma sono state descritte anche mutazioni dominanti, tuttavia rare.
Le condizioni β0 o β+ non sono correlabili alla gravità della talassemia. In genere, tanto la talassemia major quanto quella intermedia sono legate a una condizione di omozigosi β0 o β+ o di eterozigosi β0β+.
È invece la tipologia di mutazioni determinanti la condizione β0 o quella β+ a incidere sulla gravità delle manifestazioni cliniche.
Nella talassemia minor saranno presenti un allele con mutazioni del tipo β0 o β+ e un allele normale.
Link correlati:
Che cos'è l'albinismo oculocutaneo?
Studia con noi