Fenilchetonuria
Che cos'è la fenilchetonuria?
La fenilchetonuria è una malattia genetica, a trasmissione autosomica recessiva, che provoca un'alterazione nella reazione di conversione dell'aminoacido fenilalanina in tirosina.
Questa reazione rappresenta la prima tappa della via di degradazione della fenilalanina, i cui livelli nel sangue risultano di conseguenza molto alti (iperfenilalaninemia).
Non esiste una vera e propria cura della fenilchetonuria, il trattamento prevede una dieta che limiti il più possibile l'apporto di fenilalanina.
L'incidenza della fenilchetonuria è variabile a seconda dei gruppi etnici, presentandosi con una frequenza media di 1 su 10.000/15.000 nati. La Finlandia ha l'incidenza minore, 1 su 100.000 nati, mentre la più alta (1 su 4.000) si riscontra in Turchia a causa dei frequenti matrimoni tra consanguinei. In Italia si registrano 50 nuovi casi ogni anno.
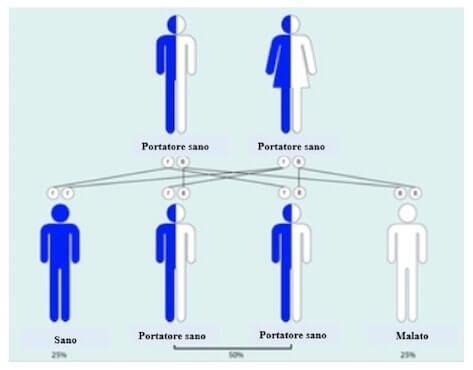
La fenilchetonuria è una malattia genetica che si trasmette in modo autosomico recessivo. In accordo con questo meccanismo di ereditarietà, la probabilità che nasca un individuo affetto da due portatori sani è del 25%.
Cause della fenilchetonuria
La fenilchetonuria è causata da mutazioni nel gene per la fenilalanina idrossilasi (PAH), situato sul braccio lungo del cromosoma 12. Questo enzima interviene nella prima tappa del catabolismo dell'aminoacido fenilalanina convertendolo in tirosina.
In assenza dell'enzima PAH, la fenilalanina va incontro a una via di degradazione alternativa che porta alla produzione di fenilpiruvato, parte del quale viene convertito in fenilacetato e fenillattato. La fenilalanina, il fenilpiruvato e i suoi derivati si accumulano nel sangue e nei tessuti e vengono escreti con le urine (da qui il nome fenilchetonuria).
Un'altra possibile causa della fenilchetonuria, ma meno frequente, è la carenza di tetraidrobiopterina (BH4), un cofattore di cui necessita la fenilalanina idrossilasi. In questi casi i livelli di fenilalanina nel sangue sono inferiori rispetto alla fenilchetonuria classica.
La tetraidrobiopterina nel corso della reazione catalizzata dalla fenilalanina idrossilasi viene ossidata a diidrobiopterina (BH2), nuovamente ridotta a BH4 dall'enzima diidrobiopterina reduttasi.
La carenza di BH4 può essere dovuta a difetti negli enzimi implicati nella sintesi o nella rigenerazione.
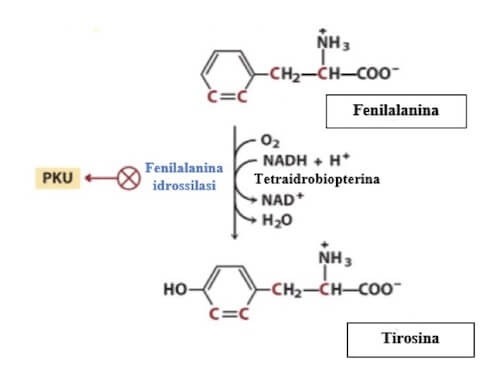
La fenilchetonuria è dovuta a difetti nell'enzima fenilalanina idrossilasi (PAH), il primo ad essere coinvolto nella via di degradazione della fenilalanina. L'enzima PAH converte la fenilalanina in tirosina, che continua nel processo di degradazione.
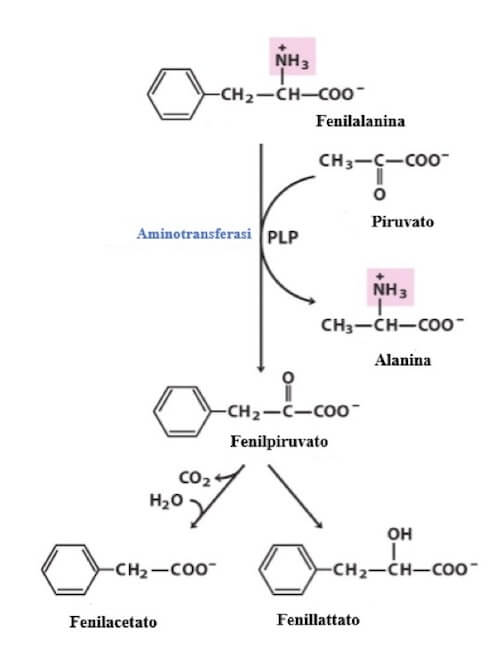
Il mancato funzionamento dell'enzima PAH attiva una via metabolica alternativa che converte la fenilalanina in fenilpiruvato. Parte del fenilpiruvato viene convertito in fenilacetato e fenilattato. La PKU si caratterizza per elevati livelli di fenilalanina e fenilpiruvato nel sangue, nei tessuti e nelle urine. Anche fenilacetato e fenillattato possono essere presenti nell'urina.
Manifestazioni cliniche della fenilchetonuria non trattata
Nei casi di fenilchetonuria non trattata, l'accumulo di fenilalanina o dei suoi derivati nei primi mesi di vita provoca uno sviluppo anomalo del cervello con conseguente ritardo mentale. L'eccesso di fenilalanina limita, inoltre, il passaggio attraverso la barriera emato-encefalica e la disponibilità al cervello di altri nutrienti essenziali.
L'alterazione delle funzionalità neuronali deriva anche dalla riduzione dei livelli di tirosina, da cui derivano importanti neurotrasmettitori, come dopamina, noradrenalina e adrenalina.
L'accumulo di fenilalanina e dei prodotti derivati dalle reazioni alternative che si attivano nei bambini affetti da fenilchetonuria provoca un odore caratteristico di "topo".
Spesso i bambini affetti hanno anche pelle e capelli di colore più chiaro rispetto agli altri componenti della famiglia.
La fenilchetonuria non trattata può provocare anche difficoltà motorie, convulsioni, dermatiti eczematose, disordini dello spettro autistico e comportamentali vari.
Diagnosi e trattamento
La fenilchetonuria può essere facilmente diagnosticata alla nascita mediante una semplice analisi al sangue e trattata sin da subito con un'alimentazione che elimini il più possibile gli alimenti ricchi di proteine.
Sono da evitare anche i cibi che contengono dolcificanti artificiali come l'aspartame, nella cui composizione chimica è presente fenilalanina.
Il trattamento della fenilchetonuria prevede, inoltre, cibi appositamente formulati, privi di fenilalanina ma non degli altri aminoacidi.
Questi alimenti specifici, tuttavia, risultano spesso poco appetibili e possono causare carenze alimentari, soprattutto delle vitamine D e B12, il cui apporto deve essere accuratamente monitorato.
Se l'alimentazione povera di fenilalanina viene adeguatamente seguita sin dalla nascita è possibile limitare e ridurre al minimo il ritardo mentale, ma alcune difficoltà fisiologiche e psicologiche possono persistere.
Link correlati:
Che cos'è la corea di Huntington?
Che cosa è la fibrosi cistica?
Studia con noi